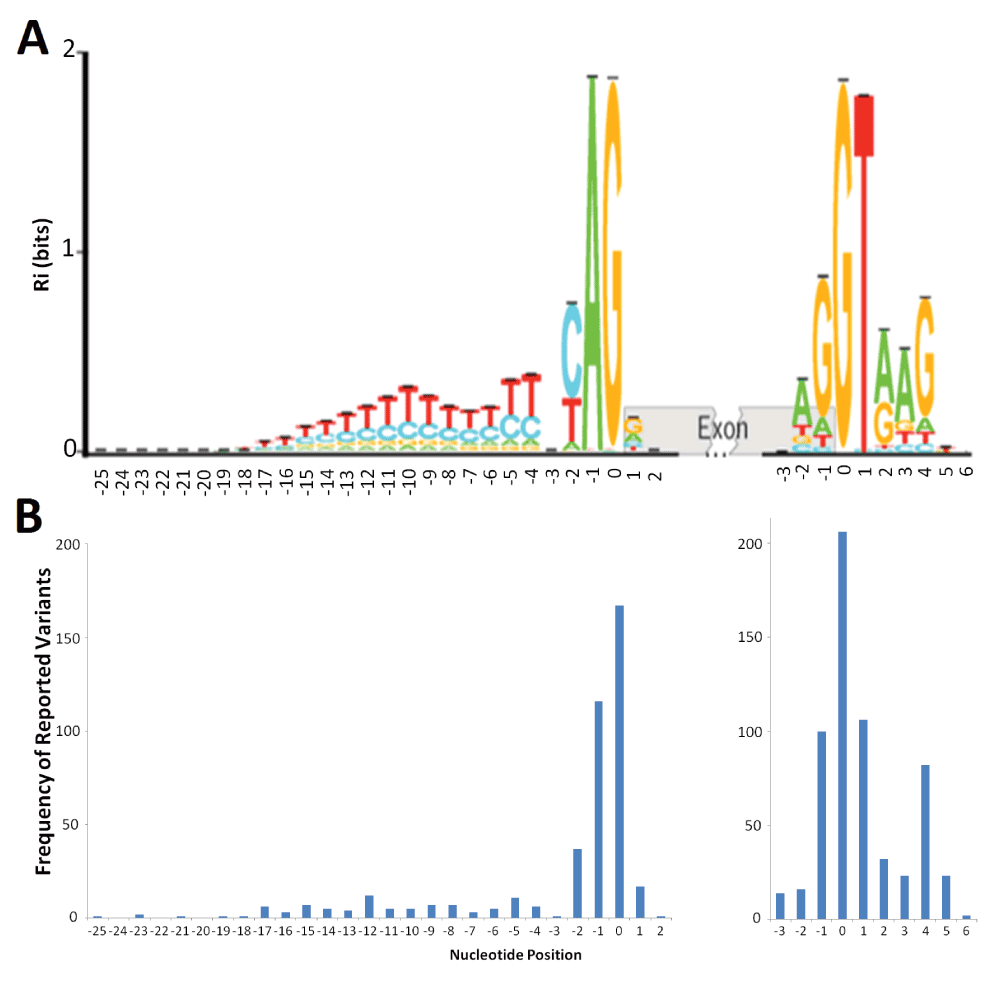
Distribution of deleterious natural site variants relative to information content.
A) The sequence logo for human acceptor and donor splice sites based on the positive (+) strand of the October 2000 (hg5) genome draft. The logo shows the distribution of information contents (Ri in bits) at each position over the region of 28 nucleotides for acceptor [-25, +2] and 10 nucleotides for donor [-3, +6] from the first nucleotide of the splice junction (position 0). Nucleotide height represents its frequency at that position. The horizontal bar atop each stack indicates the standard deviation at that position. This figure was modified from Rogan et al. (2003) to include splice sites in genes on both strands of the annotated human reference genome33. B) The distribution of deleterious single-nucleotide variants reported at the natural acceptor (left) and donor (right) splice sites. The variants used to populate this graph were included only if they were reported to negatively affect splicing (N = 431 for acceptors, 604 for donors). The image was aligned to the sequence logo (A) to illustrate potential correlation of number of splicing variants at a position to the information content at that position.
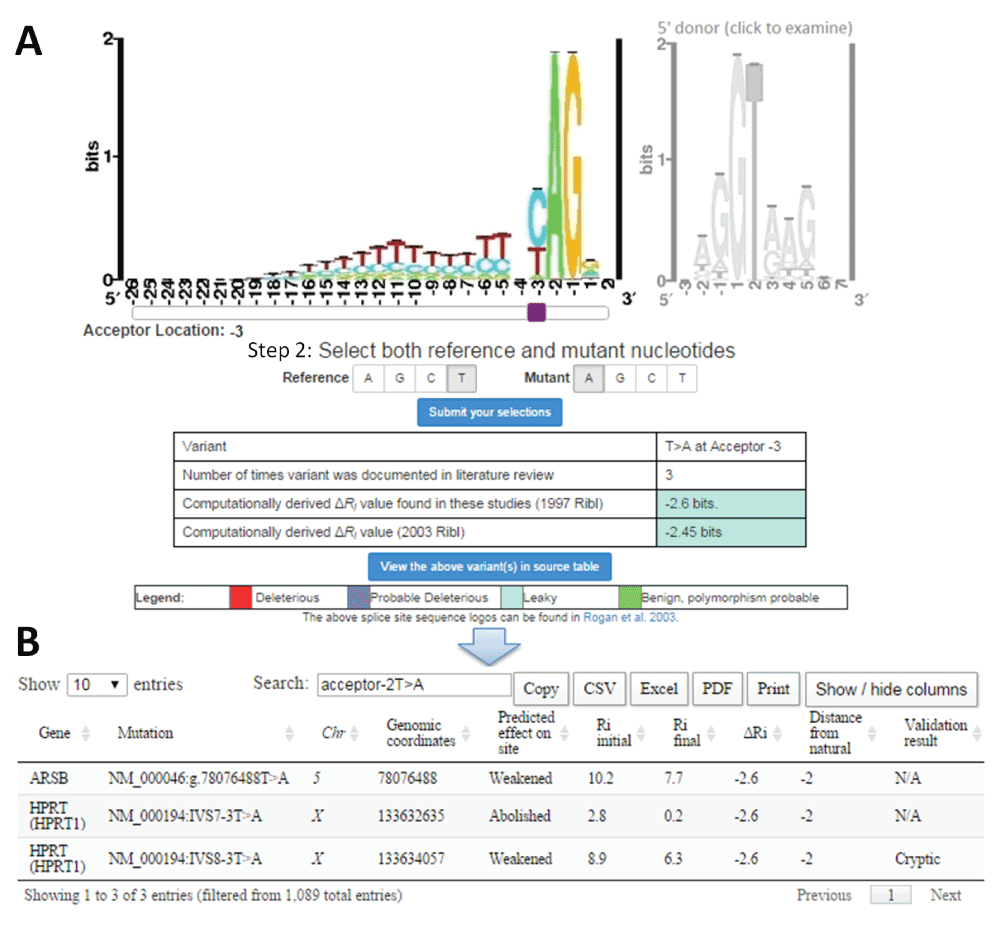
Sample retrieval of average change in information content (ΔRi) with splicing mutation calculator (SMC) for published mutations.
A) Example mutation input for SMC (T>A at the 3rd intronic position of natural acceptor). The type of splice site is selected by clicking on the corresponding sequence logo (acceptor [left] or donor [right]). The purple slider bar appearing below the logo is used to select the position of the mutation. The reference and mutant nucleotides are then designated, and the variant is submitted to the software (‘Submit your selection’). SMC outputs a table indicating the user input, the number of instances in the literature where this substitution has been analyzed using IT, and the computed ΔRi values (in bits) using both the old (1992; top) and new (2003; bottom) ribls. The cell colour for ΔRi values indicates the predicted severity of the inputted variant according to defined thresholds25,157. B) Tabular output detailing each instance of the selected mutation from the source table. The user may view, in a separate window, extensive details of all variants referred to in SMC output.
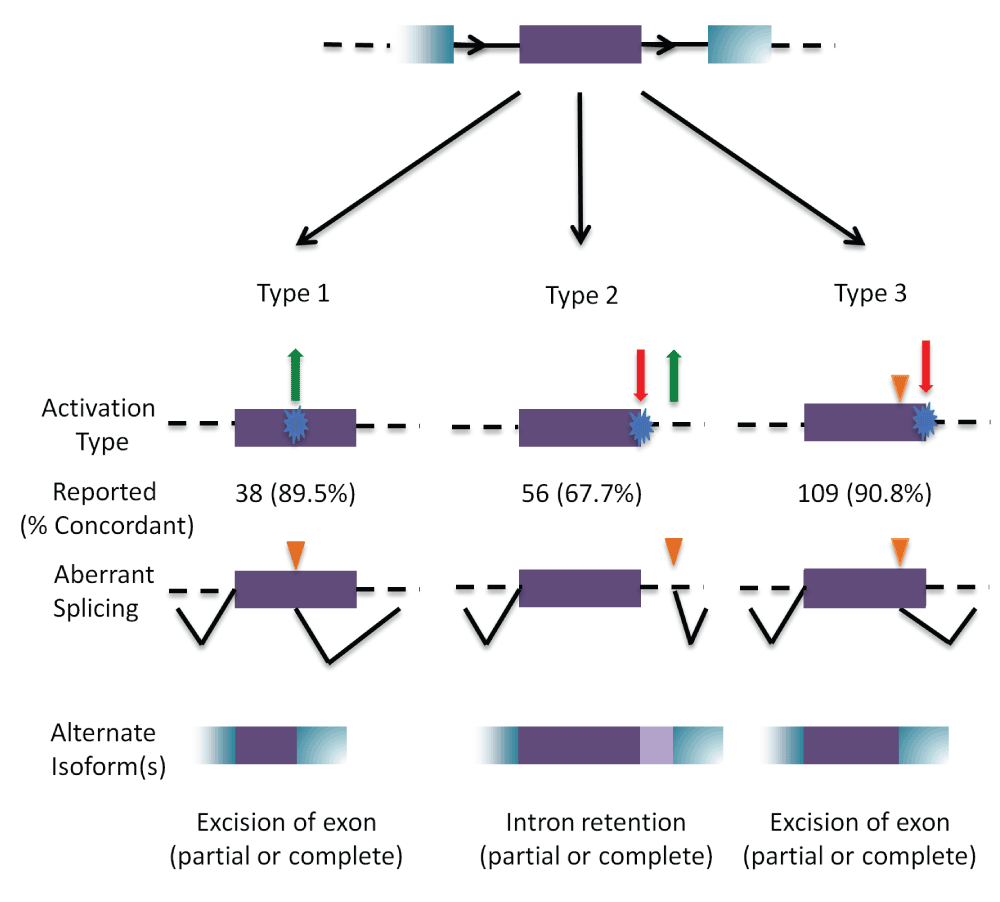
Outcomes of cryptic splicing mutations.
A prototypical internal exon (in purple) with flanking exons (in blue); introns are represented by black solid, and dashed lines (top). The three types of cryptic splice site activation are then illustrated. Type 1 cryptic splice site activation (left) is caused by the activation (green arrow) of a cryptic site by strengthening a pre-existing site, or by creating a novel splice site (blue). Type 2 (middle) results from the simultaneous weakening or abolition (red arrow) of the natural splice site while strengthening or creating (green arrow) a cryptic site. Type 3 (right) involves the activation of a pre-existing cryptic site due to the weakening or abolition of the natural splice site (indicated by orange triangle). The number of cases that have been reported in the literature that has been analyzed by IT for each type is indicated, with the percent accuracy in parentheses. The bottom row represents the resulting mRNA structure due to the activated cryptic splice site.